Jesteś tutaj


The Youngest Hero: How a Newborn Can Change the Life of an Older Sibling with Sickle Cell Disease
 What is Sickle Cell Disease?
What is Sickle Cell Disease?
Sickle Cell Disease (SCD) is a group of inherited red blood cell disorders, known as hemoglobinopathies, affecting mostly individuals of African or Hispanic descent. Common symptoms of SCD are recurrent debilitating pain, organ damage, and infection. These symptoms are caused by unhealthy red blood cells that are sickle-shaped, causing them to stick in small blood vessels. Other complications of SCD can include acute chest syndrome and neurologic complications such as stroke. People affected with SCD have shortened life expectancy, with median lifespan currently in the 40’s in the United States.
How is SCD inherited?
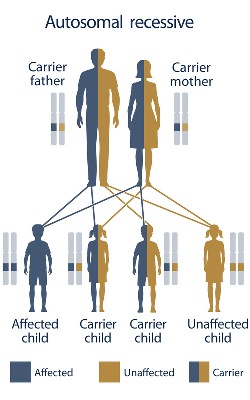
SCD is a genetic condition that only occurs when two non-working copies of the gene are inherited: one from mom and one from dad. This is called autosomal recessive inheritance. Carriers of SCD, also known as sickle cell trait, typically do not show any symptoms of the condition and do not develop related health problems. When both parents are carriers of sickle cell disease, there is a 1 in 4 (25%) chance with each pregnancy to have a child with SCD. Males and females are equally affected by SCD.
 How does a family know if they may be at risk to have a child with SCD?
How does a family know if they may be at risk to have a child with SCD?
There are two main tests that can help families know if they are at risk to have a baby with SCD: carrier screening and newborn screening.
Carrier screening on parents can be performed prior to conception or in the early stages of pregnancy. Carrier screening is performed through a blood draw and can test for hemoglobinopathies as well as other genetic conditions. Screening prior to conception allows families in which both parents are carriers more options to lower the risks of having a child with SCD.
Carrier screening during pregnancy allows at risk couples the option to test the unborn baby for SCD. These tests (amniocentesis or chorionic villi sampling) can determine if an unborn baby has SCD or is unaffected.
Newborn screening (NBS) is a test performed in every US state to screen newborn infants for a variety of conditions, including sickle cell disease. NBS is performed by a heel-prick of the newborn and results are reported to the child’s pediatrician typically within one week.
What are the treatments and/or cure for SCD?
Treatments for SCD are designed to relieve pain and prevent infections and stroke. These treatments include blood transfusions, IV-fluid therapy, and pain/infection medications. Hospitalizations are common in individuals with SCD. Severe SCD is often treated with hydroxyurea, a medication shown to reduce the number of hospital stays and blood transfusions needed. However, even with the best monitoring and treatment, severe complications of SCD can occur and life expectancy is reduced.
To date, the only cure for SCD is a transplant of blood-forming (hematopoietic) stem cells. The sources of these stem cells include: bone marrow, peripheral blood, or umbilical cord blood. Stem cell donors and recipients must be a close genetic match for one another, known as HLA matching. Siblings have the highest chance to be this type of genetic match. People undergoing a successful transplant can expect to live much longer and symptom-free. For this reason, stem cell transplants are increasingly considered in younger children and young adults with SCD.
Cord blood is underutilized as a resource for stem cell transplants. Cord blood is easily available and a rich source of hematopoietic stem cells. Because of their immaturity, cord blood stem cells do not have to be an exact HLA match and have a lower incidence of rejection. However, cord blood stem cell units may have a smaller volume of cells, resulting in longer engraftment time or the need for a double cord blood unit transplant.
How can cord blood banks help?
Based on average family sizes, an estimated 30% of individuals will have a perfect genetically matched sibling donor, but most of these individuals will not have stored cord blood stem cells in a family bank. More education about cord blood banking can increase the number of patients benefiting from cord blood stem cell transplants for conditions such as SCD. Being aware of the benefits of banking cord blood from healthy siblings empowers families to consider wider treatment options.
Evercord, a family cord blood bank powered by Natera, offers families that have a child affected with a disease treated by stem cell transplant to store another unaffected child’s cord blood at no charge. This includes families that have a child with SCD. Shaun Ford* was diagnosed with SCD at birth through newborn screening and has suffered complications over his short life of 4 years. Shaun’s parents are expecting another baby and plan to store cord blood stem cells from their new baby. Shaun’s sibling has the chance to be the youngest hero for his big brother with a 25% chance of being an exact genetic match for a stem cell transplant. (Follow up: CBR® acquired Evercord™ newborn stem cell business from Natera in Sept. 2019)
*Changed to protect patient identity
 Beth Buehler, MS CGC, is a board certified genetic counselor with over 27 years of experience in prenatal and pediatric genetics. She has been a private practice genetic counselor for 17 years working with a high risk perinatal practice in Palm Beach County Florida. Prior to that, she was at the University of Miami specializing in pediatric, prenatal and Children's Medical Service clinics. Beth is a member of the National Society of Genetic Counselors, The American College of Medical Genetics, and The American Board of Genetic Counseling. She earned her BA from the University of Florida and her MS from Sarah Lawrence College.
Beth Buehler, MS CGC, is a board certified genetic counselor with over 27 years of experience in prenatal and pediatric genetics. She has been a private practice genetic counselor for 17 years working with a high risk perinatal practice in Palm Beach County Florida. Prior to that, she was at the University of Miami specializing in pediatric, prenatal and Children's Medical Service clinics. Beth is a member of the National Society of Genetic Counselors, The American College of Medical Genetics, and The American Board of Genetic Counseling. She earned her BA from the University of Florida and her MS from Sarah Lawrence College.
References:
- Sickle Cell Disease by MA Bender, MD PhD
- Be The Match > Transplant Basics > Cord blood and transplants
- Walters MC, De Castro LM, Sullivan KM, et al. Indications and results of HLA-identical sibling hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2016; 22(2): 207-211
- Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: Indications and management recommendations from an international expert panel. Haematologica 2014; 99(5): 811-820.
- National Cord Blood Program > cord Blood Q&A