当前位置


Role of Cord Blood Transplants to Treat Sickle Cell Anemia & Thalassemia in the Age of Gene Therapy
In recent years, multiple new therapies have appeared for the treatment of Sickle Cell Anemia and Thalassemia. This leads to a legitimate question: Do cord blood transplants still have a role to play in the treatment of these diseases? The bottom-line-up-front answer is YES, cord blood transplants will continue to be valuable as a cure for the foreseeable future.
Sickle Cell Anemia and Thalassemia are both “hemoglobinopathies”: they are diseases in which the patient’s body does not properly make the correct type of hemoglobin, the key ingredient in red blood cells that enables them to carry oxygen1. In Thalassemia, the primary symptoms of the defective blood cells are anemia and infections, which cause patients to be chronically fatigued2,3. There are many variants of Thalassemia, but it is mostly the version Beta Thalassemia Major that causes patients to require many blood transfusions, which leads to organ damage from iron overload and various complications1-3. In Sickle Cell Anemia, the red blood cells are sickle-shaped instead of round1,4. In addition to experiencing anemia, Sickle Cell Anemia patients have repeated pain crises when the sickle-shaped blood cells stick together and block blood vessels4. This can cause a stroke, and over time the repeated blockages cause organ damage4.
All the hemoglobinopathies are hereditary diseases that are carried by genetic mutations1-4. As a result, they run in families, they are more prevalent in certain ethnic groups, and they are more prevalent in parts of the world where those people live. See the maps embedded in this page which display the world regions afflicted by high rates of Sickle Cell Anemia or Thalassemia5,6. When a person has only one disease-carrying genetic mutation, they are a “carrier” of Sickle Cell or Thalassemia, and their symptoms are manageable1-4. Many carriers are totally asymptomatic. But when a person inherits the genetic mutation from both parents, they will have the diagnosis of Sickle Cell Anemia or Beta Thalassemia Major1-4. It is also possible for a patient to have a crossover disease with one Sickle Cell mutation and one Thalassemia mutation.
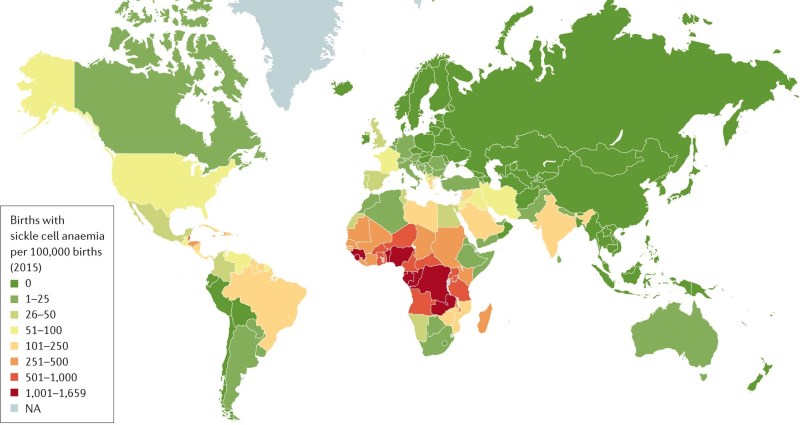
From a global perspective these are not rare diseases. The epicenter of Sickle Cell Anemia is in central Africa; in some parts of Kenya about 20% of the population carries the trait and up to 3% of births have the disease5. Without modern medicine, 50% to 90% of the children in Africa that are born with Sickle Cell Anemia will die before the age of five7. Yet ironically, adults who carry Sickle Cell trait on only one gene have much better chances of surviving Malaria8! Clearly, there is an evolutionary advantage to being a carrier of this disease, so it makes sense that Sickle Cell persists in tropical parts of the world. Over the past century, migration has led people carrying Sickle Cell trait to disperse across the world, and today the genetic testing company 23andMe estimates that 300 million people worldwide are carriers9.
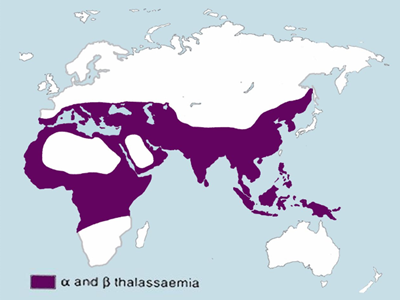 Thalassemia is most prevalent in Mediterranean countries and in Southeast Asia6,10. In the region encompassing Vietnam, Cambodia, Laos, Thailand and Malaysia, the prevalence of Thalassemia carriers is between 20% and 50% of the population10. Like Sickle Cell carriers, Thalassemia carriers also have better chances of surviving Malaria11. Thalassemia is especially common in those religious and ethnic groups where cousins marry cousins; these are called “consanguineous marriages”12. But several countries that require carrier screening prior to marriage have succeeded in dramatically lowering the birth rate of children with Beta Thalassemia Major, up until recent influxes of unscreened refugees12.
Thalassemia is most prevalent in Mediterranean countries and in Southeast Asia6,10. In the region encompassing Vietnam, Cambodia, Laos, Thailand and Malaysia, the prevalence of Thalassemia carriers is between 20% and 50% of the population10. Like Sickle Cell carriers, Thalassemia carriers also have better chances of surviving Malaria11. Thalassemia is especially common in those religious and ethnic groups where cousins marry cousins; these are called “consanguineous marriages”12. But several countries that require carrier screening prior to marriage have succeeded in dramatically lowering the birth rate of children with Beta Thalassemia Major, up until recent influxes of unscreened refugees12.
Together, about 7% of the world population is a carrier of a hemoglobinopathy, making these conditions as a group the most common genetic disorder in the world and one of the world’s major health problems13.
Hydroxyurea is a simple and effective drug treatment for Sickle Cell Anemia. First approved by the US FDA in 1967 as a treatment for blood cancers, it was discovered in the 1980’s that hydroxyurea alters the production of blood cells in such a way that fewer sickle cells are produced and hence patients have fewer blockages of their blood vessels. With a simple daily regimen of hydroxyurea, children born with Sickle Cell Anemia have over 80% survival at age five14,15. Moreover, this drug is not under patent protection anymore so it is available as a relatively cheap generic. Yet, hydroxyurea is out of reach for Sickle Cell patients in impoverished parts of Africa. For example, a specialty clinic in Kenya for sickle cell patients reports charging 20 Kenya shillings (14 cents in U$D) for a hydroxyurea pill, but even this low price can be challenging for a parent with a daily income of 300 Kenya shillings and several children to be treated16.
Since 2017, the United States FDA has approved three more drugs for managing Sickle Cell Anemia: Endari from Emmaus Medical, Adakveo from Novartis, and Oxbryta from Global Blood Therapeutics17-19. These drugs try to correct the behavior of the sickle shaped red blood cells, by making them more flexible, more slippery, and better able to hold oxygen. Without examining the relative efficacy of these drugs, the problem they have as a group is that they are expensive, even for patients in Western countries who obtain them through insurance plans. The wholesale cost per year is U$D 30,046 for Endari, U$D 96,354 for Adakveo, and U$D 92,584 for Oxbryta20. Within the United States, a non-profit organization named the Institute for Clinical and Economic Review (ICER) is relied upon as a gold standard for evaluating how the financial cost of a therapy compares to its benefit in terms of quality-adjusted life-years (QALYs). According to a 2020 report from ICER, all three of the new Sickle Cell drugs are over-priced by a factor of three to nine20,21.
Unfortunately, there are no drugs approved that correct the function of blood cells in patients with Thalassemia. However, improved management of iron overload from blood transfusions has enabled children with Beta Thalassemia Major to live longer and healthier lives12,22.
A stem cell transplant is a one-time treatment that can cure Sickle Cell Anemia or Thalassemia. However, stem cell transplants carry a small risk that the patient may die of a treatment related mortality, and some fraction of patients will develop draft versus host disease (GvHD) post-transplant and be dependent on immunosuppressive drugs23,24. Cord blood transplants have an advantage over bone marrow transplants because they cause less GvHD. Pediatric patients that have experienced less organ damage from prolonged illness with hemoglobinopathies and less iron overload from repeated transfusions have better transplant outcomes. And pediatric patients are small enough to rely on cord blood as the graft source. Hence children with a “severe” hemoglobinopathy are considered good candidates for cord blood transplants23-25. For Sickle Cell this includes patients that have had a stroke, that have acute chest syndrome, that have recurrent pain crises despite medication, or other serious complications that make the benefits of the transplant outweigh the risks. Similarly, a transplant is recommended for Thalassemia patients that are transfusion-dependent.
In the United States, the first Sickle Cell patient to have a cord blood transplant was Keone Penn in 1998. Since then, several US-based Sickle Cell patients have become advocates for cord blood transplants. Marriam Carol Mulumba had a cord blood transplant at age 7 in 2008, and has been counseling other children going through transplant ever since. She published her life story at age 12 and again at age 17. In 2023, beauty pageant contestant Sosa Evbuomwan shared that she was the first child cured of Sickle Cell Anemia by a transplant of the expanded cord blood product from Gamida Cell.
In India, about 12 to 14 thousand children are born each year with the severe form of Thalassemia26. Pediatric hematologists in India report that hemoglobinopathies are the most common indication (at 34%) for children receiving stem cell transplants27. LifeCell, the largest cord blood bank in India, notes that the majority of cord blood units they have released were for sibling transplants to treat Thalassemia26. Another study by hematologists reports that the typical cost for a sibling cord blood transplant in India is INR 20 lakhs, which is equivalent to U$D 25,00028. The costs of treatment in India are so much less than the United States that the entire cost of a cord blood transplant is less than the cost to acquire a cord blood unit from a public bank in the United States28,29.
In Iran, it was estimated in 2018 that 4% of the population were carriers of Beta Thalassemia and 18,783 patients suffering from Beta Thalassemia Major had been identified30. A 2015 survey of the economic burden of the disease found that the average annual cost to support these patients with blood transfusions, iron chelation, etc. was U$D 8321.8031. This includes the indirect burden of lost wages, but the majority of the cost, U$D 7286.80, is direct medical expenses which under the Iranian public health system are an annual cost to society of over U$D 136 million for all the patients in the country31. Royan Stem Cell Technology Company, the leading private and public cord blood bank in Iran, often provides sibling cord blood transplants for Thalassemia. The cost to privately store cord blood with Royan is about U$D 250 and the cost of the cord blood transplant is about U$D 2500. This is another example of a country where a stem cell transplant is feasible and much more cost effective than lifelong supportive care of Thalassemia.
In China, the public health burden of Thalassemia is compensated by the large inventory of donated cord blood in public banks, so that a matched donor can be found for transplant when a sibling is not available. At the 2023 cord blood conference of the China Maternal and Child Health Association, it was said that one out of six people in the southern Chinese province of Guangdong is a carrier of Thalassemia32. Professor Liu Sixi, chief physician of the Hematology and Oncology Department of Shenzhen Children's Hospital, said, "In 11 years, we performed 909 cases of Thalassemia transplantation, and the overall survival rate was 99%. This data is ahead of countries and regions such as Europe and the United States32."
In Thailand, close to the world’s epicenter for Thalassemia carriers, families are turning to assisted reproduction to help them conceive a savior sibling that can provide a cord blood transplant to an older child with Thalassemia33. Relying on pure luck, the probability that the parents will conceive a child that is an exact HLA match to their sibling is 25%, while the probability that the new baby will also carry Thalassemia is 75%. Whereas, by relying on in-vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD), the parents can guarantee conceiving a child that is both a perfect donor and not a carrier of Thalassemia. The Superior A.R.T. and Jetanin fertility centers in Thailand, which have the largest case series in the Far East and Pacific, report a live birth rate of 44% with a total cost of U$D 10,000 to 30,000, including delivery of the baby34.
In Kenya, the Swiss cord blood bank CordSavings has entered a private-public partnership with local authorities for public health in geographic areas where HLA diversity appears to be particularly high. Their joint goal is to provide worldwide Sickle Cell patients with access to better matching cord blood units for transplants. During the first half of 2023, they succeeded in commencing collection, processing, and storage of cord blood units from a large public hospital. But, as of today this effort has been temporarily put on hold due to the current civil unrest in in Kenya35.
Disease | Therapy | Manufacturer | US FDA Approved? |
Sickle Cell | Drug Hydroxyurea | Generic | Yes |
Sickle Cell or Thalassemia | Cord Blood Transplant | Any Cord Blood Bank | Yes |
Sickle Cell | Drug Endari (chemical L-glutamine) | Emmaus | Yes 2017 |
Sickle Cell | Drug Adakveo (crizanlizumab) | Novartis | Yes 2019 |
Sickle Cell | Drug Oxbryta (voxelotor) | Pfizer | Yes 2021 |
Thalassemia | Zynteglo: Gene editing of the patient’s cells outside the body | Bluebird Bio | Yes 2022 |
Sickle Cell | Lyfgenia: Lentivirus gene editing of the patient’s cells outside the body | Bluebird Bio | Yes 2023-12-08 |
Sickle Cell or Thalassemia | Casgevy (formerly Exa-cel): CRISPR gene editing of the patient’s cells outside the body | CRISPR/Vertex | Yes 2023-12-08 |
Sickle Cell | Gene editing of the patient’s cells within the body | Novartis | Research |
Gene therapy is the latest medical tool to treat patients with severe hemoglobinopathies. Sickle Cell Anemia and Beta Thalassemia are prime candidates for gene therapy because they are both “monogenic” diseases, where the genetic mutation that causes the disease is on a single gene – if you can fix that gene, you can cure the disease1.
The United States FDA approved a gene therapy for Thalassemia in August 202236, and it is widely anticipated that two gene therapies for Sickle Cell Anemia may be approved in 202337. So far, these gene therapies work by taking stem cells from the patient, genetically editing them in a laboratory, and then giving them back to the patient after some chemotherapy to wipe out the patient’s existing stem cells and make sure that the newly edited stem cells take over. This is basically giving the patient an autologous transplant of their own stem cells, with a genetic manipulation process in addition. This is more complicated than a stem cell transplant, and it is also much more expensive.
The RAND Corporation estimates that the average cost of a cord blood transplant in the United States in 2017 was U$D 831K, including a hospital stay of seven weeks29. The US price tag of the approved gene therapy for Thalassemia is more than 2.5 times higher, at U$D 2.1 million for the product alone38. The Institute for Clinical and Economic Review (ICER) approved of the $2.1 million price, by comparison to the cost of supportive care and the impact on quality of life, because the gene therapy is a one-time curative treatment38. In the phase 3 trial of the gene therapy, 90% of the Thalassemia patients achieved transfusion independence39,40. But, let us remember that cord blood transplant is also a one-time curative treatment, and there are reports of patient series with 97% to 100% success24,32,34. This is not intended to disparage gene therapy, but there is much we do not know yet about the head-to-head comparison of gene therapy versus traditional stem cell transplants for severe hemoglobinopathies. Some of the issues to be examined are the length of hospital stay, the engraftment rate, and the clinical outcomes for matched patient cohorts.
The face of gene therapy for Sickle Cell Anemia belongs to Victoria Gray, a mother of four from Mississippi who was the first person to receive CRISPR gene editing in July 2019 as part of a clinical trial. Her experiences have been chronicled by National Public Radio (NPR) in a series of exclusive interviews every few months. In December of 2021 she told NPR, "This is major for me and my family" … "Two years without me being in the hospital? Wow. We just can't believe it. But we're so grateful." In March of 2023 she was invited to speak at a conference in London about human genome editing. While sightseeing in Trafalgar Square she said, "I would never have been able to walk this long before" … "It's a huge difference — night and day. I feel like I got a second chance."
The race has already begun to develop the next generation of gene therapies for Sickle Cell Anemia and Beta Thalassemia. Multiple clinical trials are employing the current technique of extracting the patient’s own stem cells and editing them to deliver a cell-based gene therapy37,41,42. Laboratory researchers are studying what are the best ways to perform the gene editing to achieve strong and consistent responses in patients43. The next frontier will be to perform the gene editing directly in the patient’s body (in vivo), without the need to extract stem cells and process them in a laboratory. Novartis is devoting their Sickle Cell research pipeline to the in vivo gene editing approach, in collaboration with the Bill & Melinda Gates Foundation44. This approach is not yet in clinical trials.
As the general public becomes aware that gene therapies are “transformative” for genetic diseases, questions are being raised about the trade-off between the high cost of these therapies versus the need for basic public health programs45. In the case of rare diseases, expensive gene therapies can be absorbed by a public health system because the number of patients is limited. For example, ICER calculated that the U$D 2.1 million price tag of Zynteglo (with 80% refund if it doesn’t work) was reasonable in the United States because there are only 1,000 to 1,500 patients in the US living with transfusion-dependent Thalassemia38. Obviously that calculation will collapse in parts pf the world where hemoglobinopathies are more prevalent and the national economy is less robust. In the case of Sickle Cell, 75% of the world’s patients live in Africa, and 95% of Sickle Cell patients live in countries on the lower half of the wealth scale46.
The Lancet Haematology Commission has devoted an entire issue of their journal, published 11 July 2023, to defining global strategies that can improve outcomes in Sickle Cell Disease46. A large part of the report deals with the need for better screening, more systematic epidemiology data, and better training of healthcare providers. However, the Commission states that their “ultimate aim is to provide access to definitive or curative therapies for every patient with sickle cell disease.” The report also reviews the current state of cell and gene therapies and the challenges that they face. The Commission adopts 12 key recommendations and a goal deadline for each (see pages 43 to 45 of the overview). In the immediate future, the number of people diagnosed with Sickle Cell is expected to increase, due to more comprehensive newborn screening. In the arena of cell and gene therapy, the Commission calls for low- and middle-income countries to expand their capacity for stem cell transplants and to provide universal HLA typing for children with Sickle Cell Anemia. The Commission asks for clinical trials of advanced cell and gene therapies to be conducted at multiple sites so that patients from less wealthy countries can participate. The Commission also requests investment in technologies that will bring advanced therapies to the point of care. Finally, the Commission seeks more creative partnerships between governments, pharmaceutical companies, and non-profits, to help realize these visions.
For the foreseeable future, cord blood transplants will continue to be the best option for pediatric patients to obtain a one-time curative treatment for Sickle Cell Anemia or Beta Thalassemia Major. It is more feasible to bring cord blood banking and transplantation to the point of care in countries with a high proportion of hemoglobinopathy patients than to wait for gene therapies to be widely available. It is notable that the Lancet Haematology Commission on Sickle Cell Disease has called for more stem cell transplant programs in developing nations; hopefully that policy statement can be leveraged to help fund such programs.
References
- Learn.Genetics. Hemoglobin Disorders. Univ. Utah
- March of Dimes. Thalassemia. Last reviewed 2017-08
- Cooley's Anemia Foundation. The Man Behind the Name: Thomas Benton Cooley, M.D. Webpage. Copyright 2023
- Sickle Cell Disease Association of America. Webpage. Copyright 2021.
- Kato GJ, et al. Sickle Cell Disease. Nature Reviews. Disease Primers. 2018; 4:18010.
- Amid A, Saliba AN, Taher AT, et al. Thalassaemia in children: from quality of care to quality of life. Archives of Disease in Childhood. 2015; 100:1051-1057.
- Grosse SD, et al. Sickle Cell Disease in Africa. A Neglected Cause of Early Childhood Mortality. Am J Preventative Med. 2011; 41(6):S398–S405.
- Williams TN, et al. Sickle Cell Trait and the Risk of Plasmodium falciparum Malaria and Other Childhood Diseases. J Infectious Diseases. 2005; 192(1):178–186.
- 23andMe. Do you know whether you’re a carrier for sickle cell disease? We can help. Webpage. Copyright 2023
- Goh LPW, Chong ETJ, Lee PC. Prevalence of Alpha(α)-Thalassemia in Southeast Asia (2010–2020): A Meta-Analysis Involving 83,674 Subjects. Int J Environ Res Public Health. 2020; 17(20):7354.
- American Society of Hematology. Malaria and Thalassemia in the Mediterranean Basin. ASH Clinical News. Published 2019.
- Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing patterns in the epidemiology of β-thalassemia. Eur J Haematology. 2020; 105:692-703.
- Kohne E. Hemoglobinopathies. Dtsch Arztebl Int. 2011; 108(31-32):532–540.
- Ersi Voskaridou E, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood 2010; 115(12):2354–2363.
- John CC, Opoka RO, Latham TS, ... Ware RE. Hydroxyurea Dose Escalation for Sickle Cell Anemia in Sub-Saharan Africa. NEJM 2020; 382:2524-2533.
- Plüss JD, Turuban P, Masamo P. Novartis’s big bet on sickle cell disease struggles to reach Kenya. SwissInfo.ch (a branch of Swiss Broadcasting Corporation SRG SSR) Published 2022-11-08
- FDA. FDA approved L-glutamine powder for the treatment of sickle cell disease. Announcement. Published 2017-07-07
- FDA. FDA approves crizanlizumab-tmca for sickle cell disease. Announcement. Published 2019-11-15
- FDA. FDA approves drug to treat sickle cell disease in patients aged 4 up to 11 years. Announcement. Published 2021-12-17
- Institute for Clinical and Economic Review (ICER). Sickle Cell Disease. Report Published 2020-03
- American Society Hematology (ASH). ICER Report Finds That Sickle Cell Drugs Are Too Expensive. ASH Clinical News. Published 2020-01-24
- Viprakasit V, Lee-Lee C, Chong QT, Lin KH, & Khuhapinant A. Iron chelation therapy in the management of thalassemia: the Asian perspectives. Intnl J Hematology 2009; 90:435–445.
- Locatelli F, Rocha V, Reed W, ... Gluckman E. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood 2003; 101(6):2137-2143.
- Locatelli F, Kabbara N, Ruggeri A, ... Walters MC. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood 2013; 122(6):1072-1078.
- Malhotra M, & Shenoy S. Umbilical cord blood as a hematopoietic stem cell source in transplantation for pediatric sickle cell disease: current challenges and strategies. 2022; Transfusion and Apheresis Science. 61(5):103554.
- LifeCell. Resources. Last updated 2023
- Bhat et al. Pediatric Hematopoietic Stem Cell Transplantation in India: A report by Indian Pediatric Hematology Oncology Group. Poster no.759 Bone Marrow Transplantation 2018; 53:711-712.
- Yadav SP & Sachdeva A. Unrelated Cord Blood Transplantation in India: Breaking the Cost Barrier. Blood 2011; 118(21):4768.
- Verter F. RAND Corporation report on Public Cord Blood Banking Industry. Parent's Guide Cord Blood Foundation Newsletter. Published 2017-11
- Pourfathollah AA & Dehshal MH. Thalassaemia in Iran: Thalassaemia prevention and blood adequacy for thalassaemia treatment. ISBT Congress Issue 2018; 14(1):pages.
- Esmaeilzadeh F, Azarkeivan A, Emamgholipour S, Sari AA, Yaseri M, Ahmadi B, Ghaffari M. Economic Burden of Thalassemia Major in Iran, 2015. J Res Health Sci. 2016; 16(3):111–115.
- Beijing Umbilical Cord Blood Hematopoietic Stem Cell Bank. 10th National Cord Blood Conference. Company News. Published 2023-05-22
- Papadopoulos KI. Marjo and Matinn defy Thalassemia with the help of THAI StemLife and Superior ART. Parent's Guide Cord Blood Foundation Newsletter. Published 2023-02
- Tiewsiri K, Manipalviratn S, Sutheesophon W, ... Papadopoulos KI. The First Asian, Single-Center Experience of Blastocyst Preimplantation Genetic Diagnosis with HLA Matching in Thailand for the Prevention of Thalassemia and Subsequent Curative Hematopoietic Stem Cell Transplantation of Twelve Affected Siblings. Biomed Research Intnl. 2020; 2020:5292090
- CNN. Demonstrators feared dead as protests flare in Kenya over tax hikes. News. Published 2023-07-13
- FDA. FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-thalassemia Who Require Regular Blood Transfusions. Summary Basis. Published 2022-08-17
- Iyer JK. Sickle cell cures are coming. African children can’t be left behind. STATnews. Published 2023-07-12
- Institute for Clinical and Economic Review (ICER). Betibeglogene Autotemcel for Beta Thalassemia: Effectiveness and Value. Report. Published 2022-07-19
- Locatelli F, Thompson AE, Kwiatkowski JL, ... Walters MC. Betibeglogene Autotemcel Gene Therapy for Non–β0/β0 Genotype β-Thalassemia. NEJM 2022; 386:415-427.
- Kwiatkowski JL, et al. Long-Term Efficacy and Safety of Betibeglogene Autotemcel Gene Therapy for the Treatment of Transfusion-Dependent β-Thalassemia: Results in Patients with up to 6 Years of Follow-up. Blood 2020; 136(S1):51-52.
- Frangoul H, Altshuler D, Cappellini MD, ... Corbacioglu S. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. NEJM 2021; 384:252-260.
- Kanter J, Walters MC, Krishnamurti L, ... Tisdale F. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. NEJM 2022; 386:617-628.
- Mayuranathan T, Newby GA, Feng R, et al. Potent and uniform fetal hemoglobin induction via base editing. Nature Genetics 2023; 55:1210–1220.
- Novartis. Sickle Cell Disease. Web page. Published 2021
- Robbins R, Nolen S, Galdieri D. A Dilemma for Governments: How to Pay for Million-Dollar Therapies. NYTimes. Published 2023-01-25
- Piel FB, et al. Defining global strategies to improve outcomes in sickle cell disease: a Lancet Haematology Commission. Lancet 2023; 10(8):e633-e686.