当前位置


Thailand leads the world in conceiving savior siblings for cord blood transplants to cure Thalassemia





Thalassemias are blood disorders passed down through families (inherited) in which the body makes an abnormal form of hemoglobin. Hemoglobin is the protein in red blood cells that carries oxygen. Thalassemia disorders result in large numbers of red blood cells being destroyed, which leads to anemia.
In South East Asia many families carry a genetic mutation for thalassemia. The prevalence of alpha-thalassemia is 40% in Northern Thailand and Laos while beta-thalassemia varies between 1-9%. The hemoglobin E (HbE) mutation also occurs in 30-40% of this population and reaches 60% prevalence in certain areas of North East India. These abnormal globin genes, present in different combinations, lead to over 60 different thalassemia syndromes. About 15 million people worldwide were sick from thalassemia in 2013, making it a public health issue in those countries with many carriers.
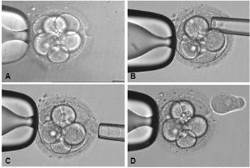
A child who has inherited hemoglobin mutations from both parents and is sick with thalassemia can only be cured by a stem cell transplant (such as a cord blood transplant). The stem cells must come from a donor who does not have thalassemia, but the donor can be a carrier of one mutation. We also know that the ideal donor for a stem cell transplant is a matched sibling. Hence, it makes sense that parents who have a child sick with thalassemia would want to genetically screen any future children. Using Preimplantation Genetic Diagnosis (PGD) with HLA matching to screen embryos before pregnancy enables the parents to conceive a child who is both healthy as well as a perfect transplant match for their sick child.
At Superior A.R.T Centre for Assisted Reproduction and Preimplantation Genetic Diagnosis , we are the leading experts in Asia helping families to conceive a "savior sibling" who can provide a cord blood transplant to an older child with thalassemia. We have achieved world-class success rates in pregnancies conceived via PGD, and we use the highest technology available to screen all 23 pairs of chromosones. We work in partnership with THAI StemLife to save cord blood from the babies and prepare it for transplantation. Below we describe the first five families that we have helped.
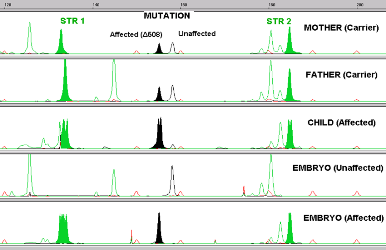
Between 2007 and 2014, 87 couples came to Superior A.R.T. seeking PGD because they were carriers of beta-thalassemia. Among them, 35 (40%) had an older child afflicted with beta-thalassemia for which they sought an HLA-matched sibling. The affected beta thalassemia children were being treated with frequent blood transfusions and iron chelation, and the parents had been informed by their children's hematologists that their only option for a cure was a stem cell transplant. The hematologists had discussed the possible sources for the stem cell transplant (bone marrow, peripheral blood, umbilical cord blood) as well their options for finding a donor in local and international registries. These families had sought an HLA-matching donor in the international registries without success.
Since they could not find a donor in the registries, these families turned to the Assisted Reproduction Technique (A.R.T.) of PGD combined with HLA matching. The goal is to select embryos which are HLA compatible with the sick child but also to select against embryos that carry thalassemia mutations, so that the resulting baby could be a stem cell donor to the sick child. All parties involved discussed the plan. This included the parents, the treating hematologists, the specialists at Superior ART (Drs. Manipalviratn and Tiewsiri) and at THAI StemLife (Dr. Fongsarun).
Finally, after 40 cycles of A.R.T., 23 of the 35 mothers (66%) had a single embryo transfer and 13 of those 23 (57%) became pregnant, while 1 miscarried. Nine babies are born to date (one set of identical twins), and in each case umbilical cord blood was successfully collected and stored with THAI StemLife.
Five of the children who were sick with beta thalassemia (three girls, aged 3, 3, and 5; and two boys, both aged 4 years) have successfully undergone cord blood transplants from their baby siblings and all are now molecularly confirmed to be cured of thalassemia. In one case cord blood stem cells from two identical twin girls were pooled and used together. In the seventh baby, the stem cell count of the cord blood was not adequate for a transplant (the affected child is 13 years old) and an appropriate age for bone marrow harvest is awaited so that the child can donate a combination of cord blood and bone marrow stem cells for transplant. The eighth and ninth babies continue to store their cord blood with THAI StemLife while waiting approval from the treating hematologist to perform their sibling's transplant.
Of the PGD-HLA babies born to date from embryo transfers, all were HLA compatible with their affected sibling. There were no misdiagnoses. Our series of cases demonstrate that directed donation of cord blood via a mutation-free and HLA-matched embryo was possible in all of the cases that sought this option, with very high pregnancy and birth rates. PGD-HLA matching is a reliable technique that provides a pragmatic and practical option for couples seeking treatment for a beta thalassemia affected child.
Cord blood transplants from PGD-HLA matched siblings are a viable alternative for families who have a child with thalassemia. The search for an HLA matched donor in the international registries failed for these 35 families seeking help. Were it not for the option to pursue A.R.T. and conceive a savior sibling, they would have been forced to pursue a transplant with a haploidentical adult donor, if they were able to pursue transplant at all. For families of Asian heritage there is the additional obstacle that they have very little chance of finding a matching adult donor outside their own ethnic group.
In the future, we hope that directed cord blood donation from PGD-HLA matched siblings will have wider applications in many more acquired and inherited pediatric disorders of the blood, immune system, and metabolism.
![]() Dr. Somjate Manipalviratn is an OB/Gyn, reproductive endocrinology and infertility specialist who graduated from the Faculty of Medicine at Chulalongkorn University, Bangkok Thailand, where he also completed his clinical fellowship in reproductive medicine. He pursued a fellowship in reproductive endocrinology and infertility with the Dept. of O&G at UCLA in Los Angeles USA, and he was a research fellow at the National Institutes of Health in Bethesda USA. Currently he is the scientific director of Superior A.R.T. but he is also based at Jetanin Inst. of ART in Bangkok Thailand. Dr. Kasorn Tiewsiri earned a PhD in molecular genetics and genetic engineering from the Institute of Molecular Biosciences at Mahidol University on scholarship from the Royal Golden Jubilee PhD Programme and the Thailand Research Fund. Following that, she pursued a 3 year biochemistry and molecular biology post-doc at Cornell in Ithaca USA. Currently she is the genetics laboratory manager at Superior A.R.T. Dr Juthatip Fongsarun is a graduate from the Faculty of Medicine at Chulalongkorn University, Bangkok Thailand, and a board certified pediatrician and pediatric hemato-oncologist. Currently she is the medical director of THAI StemLife.
Dr. Somjate Manipalviratn is an OB/Gyn, reproductive endocrinology and infertility specialist who graduated from the Faculty of Medicine at Chulalongkorn University, Bangkok Thailand, where he also completed his clinical fellowship in reproductive medicine. He pursued a fellowship in reproductive endocrinology and infertility with the Dept. of O&G at UCLA in Los Angeles USA, and he was a research fellow at the National Institutes of Health in Bethesda USA. Currently he is the scientific director of Superior A.R.T. but he is also based at Jetanin Inst. of ART in Bangkok Thailand. Dr. Kasorn Tiewsiri earned a PhD in molecular genetics and genetic engineering from the Institute of Molecular Biosciences at Mahidol University on scholarship from the Royal Golden Jubilee PhD Programme and the Thailand Research Fund. Following that, she pursued a 3 year biochemistry and molecular biology post-doc at Cornell in Ithaca USA. Currently she is the genetics laboratory manager at Superior A.R.T. Dr Juthatip Fongsarun is a graduate from the Faculty of Medicine at Chulalongkorn University, Bangkok Thailand, and a board certified pediatrician and pediatric hemato-oncologist. Currently she is the medical director of THAI StemLife.![]() Dr. Konstantinos Papadopoulos is an MD with specialty in endocrinology and diabetes, and also holds a PhD in endocrinology and immunology from University of Lund in Sweden where he has held clinical and academic positions. He has also been a lecturer in endocrinology and diabetes at the University of Alexandroupolis, Greece. At present he is the chief of medical research & development at THAI StemLife, an international biotechnology company specializing in stem cell storage and clinical applications.
Dr. Konstantinos Papadopoulos is an MD with specialty in endocrinology and diabetes, and also holds a PhD in endocrinology and immunology from University of Lund in Sweden where he has held clinical and academic positions. He has also been a lecturer in endocrinology and diabetes at the University of Alexandroupolis, Greece. At present he is the chief of medical research & development at THAI StemLife, an international biotechnology company specializing in stem cell storage and clinical applications.